「科研干货」空间分布函数SDF的计算及三维密度函数示意图
编辑:admin
来源:兰越手游网
2023-06-06
:分子动力学模拟中常以径向分布函数(radical distribution function, RDF)来描述粒子周围环境的分布特性, 并表征其短程有序性. 分布函数的概念可推广到三维, 三维空间的分布函数被称为空间分布函数(spatial distribution function, SDF), 可定义为与粒子的局部数密度与平均数密度之比,数学实质是三维的某种密度分布。一般来讲, 三维密度函数的图示方法有好几种, 其中主要的几种在量子化学的轨道图示中都有使用过, 如点云图, 径向分布图, 角度分布图, 等值面图, 切面图. 此外还有几种图示方法是计算流体力学中常用的, 在化学中使用较少.计算方法SDF计算过程与具体体系相关, 不易统一, Gromacs提供了一个g_spatial的工具, 但使用不是很方便. 很多人使用gOpenMol, 如 Distrubutions of Counterions around DNA. Molecular Dynamics Simulations Results. 我没有使用过gOpenMol, 无法评论.下面是SDF的计算方法中心分子: 确定要分析的目标分子PBC平移: 将每一个目标分子平移至原点作为中心, 根据周期性边界条件(periodic boundary condition, PBC)对其他分子进行相应平移最佳叠合/旋转一致: 对PBC平移后的构型进行最佳叠合或旋转, 使中心分子取向一致统计密度: 利用中心分子取向一致后的构型统计其周围粒子的密度分布几点说明PBC平移的方法可参考我的博文取整截断函数及其在PBC中的使用。若每帧轨迹中含有N个目标分子, 则每帧轨迹最多可得到N个构型. 使用每个目标分子作为中心分子是为了充分利用每帧的数据, 如果你的轨迹文件时间很长, 每帧只使用一个中心分子也可以。最佳叠合/旋转一致过程我以前也简略说过. 如果中心分子在MD过程中构型变化不大, 或其关键部分是刚性的, 变化不大, 那么只要选择中心分子上不共线的三个原子, 第一个原子作原点, 第二个原子处于x轴正向, 第三个原子处于xy平面, z轴垂直于三个原子所在的平面, 将所有PBC平移后的构型都旋转至标准取向即可。严格来说, 应该对中心分子做最佳叠合, 使所有中心分子相对于参考中心分子的位置的偏差(RMSD)最小. 这不是一个简单的工作, 但可利用四元数方法解决。多个分子的叠合显示可利用Jmol或VMD. VMD支持的多帧XYZ文件, 原子类型及数目必须固定, 有时使用不便; Jmol支持的XYZ文件更宽泛, 但无法操控太大的文件。VMD处理大文件的能力很赞, 一百万个原子轻松, 再多点稍困难. 我最大操作四百万个原子, 比较卡了。统计密度时, 一般是使用直角坐标, 将整个空间分为若干均匀的小格子, 然后统计每个小格子中出现的原子个数, 再除以小格子的体积, 平均, 得到原子的数密度, 将此密度与平均密度相比, 得到SDF。统计密度时, 可只统计中心分子附近某一区域内的(可由RDF获知), 因离中心分子很远的地方SDF基本趋于均匀, 意义不大。一般需要对多帧轨迹进行计算统计, 直至算出的SDF数值收敛。一般将SDF保存为cube文件. cube文件格式始于Gaussian, 但现在已基本成为标准格式了。cube文件的可视化程序很多, 大凡好点的分子可视化程序都支持, 常用的如GaussView, VMD, Jmol, ChemCraft, Chemirea, gOpenmol. 如果要同时显示多个等值面, GaussView可能不是一个好的选择, VMD可能最简单, 具体方法可参考 VMD 画cube文件等密度面图(轨道或电子密度等)代码CntAtmRotMolSDF示例如下:以一帧含5000个水分子的构型为例, 计算水分子周围O原子的SDF目标分子, 水分子, 此帧5000个2.以每个水分子为中心, 其余水分子PBC平移至[-L/2, L/2]范围内, 共得5000个构型. 其中一个如下图3.将中心水分子进行叠加显示, 可见最佳叠合/旋转一致前, 由于取向随机, 中心O原子周围H原子基本成球状分布, 中心水分子周围其他水分子分布也呈均匀分布4.将中心水分子旋转一致后, 中心O原子周围H原子分布与单个水分子类似, 但由于水分子构型在MD过程中有变化, 不会完全重合5.旋转一致后, 中心水分子周围其他水分子的分布不再均匀, 显示出各向异性, 这种各向异性从叠合后的构型图也可看出,O原子分布。H原子分布水分子分布6.计算cube文件, 利用VMD显示等值面, 两个不同值的等值面(蓝色为5, 红色为10)如下, 水分子周围H氢键的四面体结构很明显如下图:
标签:
相关游戏








相关文章
-

濡沫江湖评测:武侠迷最爱的一款游
濡沫江湖评测:武侠迷最爱的一款游戏[多图],濡沫江湖好不好玩?这是大家最喜欢的中国武侠RPG游戏,门派、副本、挖宝等各种玩法应有尽有,说实话这款游戏还是挺好玩的,不过
-

帝王上阵有讲究?这是《空岛幻想》
大家在斯特尔研究院指挥帝王上阵杀敌的时候,会怎么来安排阵容呢?
-

封龙战纪好玩吗?最新游戏评测解析
封龙战纪好玩吗?最新游戏评测解析[视频][多图],封龙战纪手游怎么样?这款游戏将会在今天正式开测,相信等待这款游戏的玩家很多,这次小编将会为大家分享封龙战纪这款游
-

11游侠评测:“突突突”超刺激射击
11游侠评测:“突突突”超刺激射击地牢游戏[多图],11游侠这款游戏好玩吗?又是一款地牢冒险手游,对于这种类型的游戏,很多玩家都会保持怀疑的状态,现在市场上的地牢游戏
-

甜甜圈都市好玩吗?一款吞噬游戏的
甜甜圈都市好玩吗?一款吞噬游戏的评测[多图],甜甜圈都市这款游戏怎么样?与其他吞噬游戏略有点不同,通关之后你会发现其实还挺有寓意的,其中一些细节非常值得深思,为了
-

星耀少女评测:二次元手游的巅峰之
星耀少女评测:二次元手游的巅峰之作[多图],星耀少女好不好玩?这款二次元游戏,从刚开始给人的感觉就是人物立绘不错,浓浓的二次元风格、精美的画质、紧凑的剧情,让这款
资讯排行
-
1
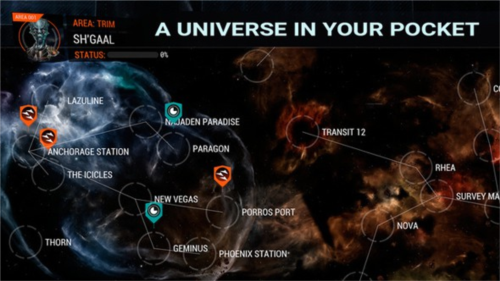
《浴火银河3》手游评测:将浩瀚宇宙展现的淋漓尽致
时间:2023-06-06
-
2

《王牌竞速》邀您成为梅赛德斯新车主
时间:2023-06-06
-
3

《地下城与勇士起源》手游评测:上线流程像被按下加速键
时间:2023-06-06
-
4

《剑与远征启程》手游评测:画风优秀玩法成熟
时间:2023-06-06
-
5
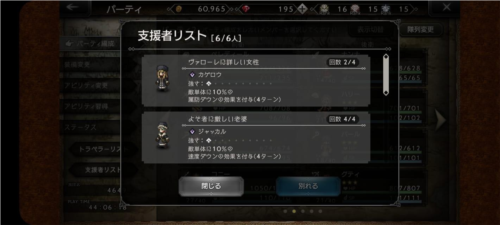
《歧路旅人大陆的霸者》手游评测:是上手了解IP的机会
时间:2023-06-06
-
6

《少年西游记2》手游评测:三真元素十分吸引人
时间:2023-06-06
-
7

《出发吧麦芬》测评:轻松好玩的护肝片
时间:2023-06-06
-
8
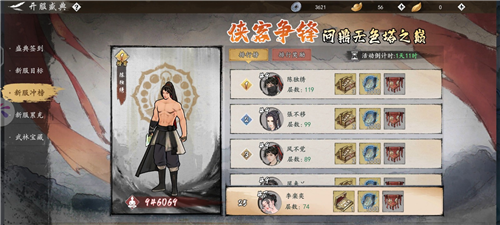
《墨剑江湖》测评:画风跟玩法都比较特殊
时间:2023-06-06
-
9

《归龙潮》手游评测:一款披着手游皮的单机
时间:2023-06-06
-
10

《余烬风暴》手游评测:沉浸美学魔幻世界
时间:2023-06-06
